Tổng quan bệnh Hội chứng Smith Lemli Opitz
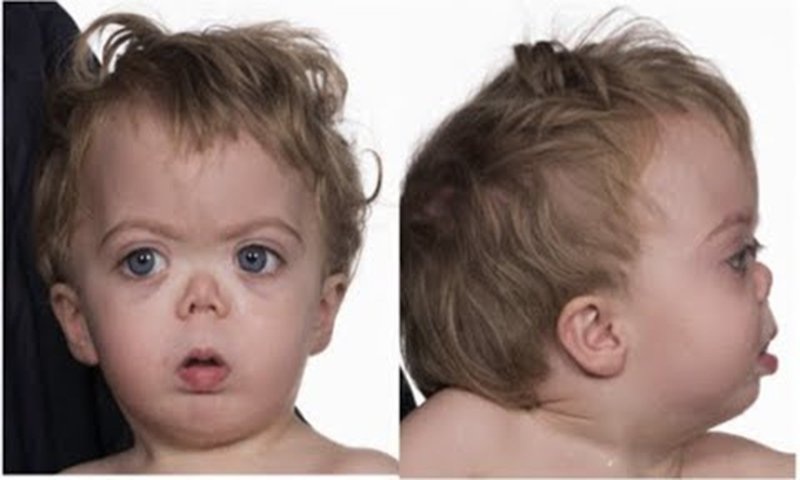
Hội chứng Smith Lemli Opitz (Smith–Lemli–Opitz syndrome – SLOS) là một rối loạn phát triển đặc trưng bởi những đặc điểm như có khuôn mặt đặc biệt, kích thước đầu nhỏ, thiểu năng trí tuệ hoặc có vấn đề trong khả năng học tập hoặc có những vấn đề về hành vi. Những đứa trẻ bị mắc phải hội chứng này thường bị tự kỷ, ảnh hưởng đến khả năng giao tiếp và tương tác xã hội của trẻ. Nó cũng thường kèm theo các dị tật ở tim, phổi, thận, đường tiêu hóa và cơ quan sinh dục. Ngoài ra, hầu hết các trường hợp mắc hội chứng này đều có dị tật vùng ngón như hợp nhất ngón chân thứ hai và thứ ba (syndactyly), và một số có ngón tay hoặc ngón chân phụ (polydactyly).
Hội chứng này được mô tả lần đầu tiên vào năm 1964, các bác sỹ nhi khoa là David Smith, Luc Lemli và Opitz John đã mô tả 3 bệnh nhân nam có biểu hiện đa dị dạng bẩm sinh và chậm phát triển tâm thần và xác định tình trạng này là RSH bằng chữ cái đầu của họ của những bệnh nhân này. Sau đó, tên của hội chứng đã được đổi thành họ của những người khám phá (SLO).
Tỷ lệ xuất hiện hội chứng Smith Lemli Opitz là khoảng 1/60.000 đến 1/20.000 ca sinh sống trên toàn thế giới. Hội chứng này dễ bắt gặp thấy ở người châu Âu, nhất là ở các nước thuộc vùng Trung Âu như Cộng hòa Séc hoặc Slovakia. Tuy nhiên, nó rất hiếm gặp ở người Châu Phi và người Châu Á.
Nguyên nhân bệnh Hội chứng Smith Lemli Opitz
Nguyên nhân gây bệnh không được biết cho đến năm 1993, Irons, Elias và Tint đã khám phá ra các bệnh nhân bị mắc hội chứng Smith Lemli Opitz có mức cholesterol máu thấp và có sự tích tụ của tiền chất sterol như 7 DHC do có sự thiếu hụt trong quá trình sinh tổng hợp cholesterol mà nguyên nhân là do hiện tượng đột biến gen gây ra. Ngày nay người ta xem hội chứng SLO là sự khiếm khuyết chuyển hoá bẩm sinh.
Hội chứng Smith Lemli Opitz là hậu quả của 1 tình trạng rối loạn di truyền (đột biến) gen DHCR7 nằm trên nhiễm sắc thể số 11, chịu trách nhiễm mã hóa quá trình sinh tổng hợp enzyme 7-dehydrocholesterol reductase (là enzyme xúc tác cho quá trình tổng hợp cholesterol) dẫn đến hậu quả làm rối loạn quá trình sinh tổng hợp cholesterol (giảm). Như ta đã biết, cholesterol là thành phần quan trọng cấu trúc nên màng tế bào cũng như là thành phần để tổng hợp nên nhiều hormone trọng yếu của cơ thể, thành phần chính cấu tạo nên vỏ myelin của sợi thần kinh. SLOS gậy đột biến gen DHCR7, từ đó làm giảm sự tổng hợp cholesterol của cơ thể, tùy mức độ thiếu hụt mà có các biểu hiện lâm sàng khác nhau, từ nhẹ với các biểu hiện thiểu năng trí tuệ, dị tật môt số cơ quan cho đến những trường hợp nặng có thể sảy thai, thai chết lưu hoặc tử vong ngay sau khi ra đời không lâu.
Đột biến gen DHCR7 là một đột biến gen lặn trên nhiễm sắc thể thường, điều đó có nghĩa người bệnh đã thừa hưởng gen đột biến gây bệnh từ cả bố và mẹ thì mới có thể có biểu hiện bệnh trên lâm sàng. Nếu chỉ nhận 1 gen bệnh từ bố hoặc mẹ thì sẽ không có biểu hiện bệnh trên thực tế, nhưng bạn sẽ trở thành người mang gen và có thể sẽ truyền lại gen gây bệnh cho con bạn trong tương lai. Trường hợp cả bố và mẹ đều mang gen gây bệnh thì 25% con của cặp bố mẹ này sẽ thừa hưởng đầy đủ cả 2 gen gây bệnh và biểu hiện thành bệnh, 50% con sẽ thừa hưởng gen đột biến từ bố hoặc mẹ và trở thành người mang gen trong tương lai; chỉ có 25% con sinh ra là có thể hoàn toàn bình thường vì hoàn toàn không thừa hưởng gen đột biến nào từ cả bố và mẹ.
Triệu chứng bệnh Hội chứng Smith Lemli Opitz
Các triệu chứng của hội chứng Smith Lemli Opitz có thể rất khác nhau tùy thuộc vào người bị ảnh hưởng, đặc biệt là tùy thuộc vào lượng cholesterol mà cơ thể có thể sản xuất ra. Các biểu hiện rất thay đổi, nếu tình trạng nhẹ, có thể chỉ có một số vấn đề trong học tập và hành vi; nhưng trong những trường hợp nghiêm trọng nhất, bệnh nhân có thể bị thiểu năng trí tuệ và những bất thường về thể chất nặng nề có thể dẫn đến tử vong. Các triệu chứng có thể xuất hiện ngay từ lúc mới sinh, cũng có thể là dần biểu hiện ra trong quá trình phát triển của người bệnh:
Chậm phát triển trí tuệ (100%)
Tự kỷ
Chậm phát triển thể chất, vận động
Rối loạn hành vi: hành vi chống đối xã hội, tự hủy hoại và bạo lực.
Chứng đầu nhỏ - Microcephaly (90%).
Các dị tật cơ xương bao gồm dư ngón tay hoặc ngón chân hoặc dính các ngón tay hoặc các ngón chân
Hở hàm ếch, hàm nhỏ
Bộ phận sinh dục ngoài của nam giới kém phát triển
Sụp mí mắt, các nếp nhăn ở mí mắt trên và dưới, mắt lác
Đục thủy tinh thể sớm
Tai lớn, nghe kém
Các vấn đề thần kinh như co giật
Giảm trương lực
Tắc nghẽn ruột
Nhạy cảm ánh sáng
Dị tật tim; thận đa nang; thiểu sản thận (1 bên hoặc cả 2 bên); tăng sản tuyến thượng thận, bệnh gan …
Hẹp môn vị, Bệnh Hirschprung (phình đại tràng bẩm sinh)
Các biện pháp chẩn đoán bệnh Hội chứng Smith Lemli Opitz
Để chẩn đoán hội chứng Smith Lemli Opitz, đầu tiên bác sĩ sẽ thu thập bệnh sử chi tiết của bệnh nhân bao gồm cả lịch sử gia đình để xác định xem có bất kỳ thành viên nào khác trong gia đình có những triệu chứng tương tự.
Những đứa trẻ ra đời mà có những dấu hiệu như lơ mơ, suy hô hấp, điếc, mù, ói mửa, khó bú, chậm lớn, táo bón, tím tái, suy tim xung huyết, nhạy cảm với ánh sáng, bất thường tâm thần kinh … thì gợi ý chẩn đoán hội chứng Smith Lemli Opitz. Khi khám xét, bác sĩ có thể phát hiện ra các dấu hiệu: đầu nhỏ, lỗ mũi rộng tẹt, hàm nhỏ, sa mi mắt, mắt bị lác, đục thủy tinh thể, dái tai thấp, dính ngón chân 2 và 3, vẻ mặt giống hội chứng Down (3 nhiễm sắc thể số 21), vòm hầu chẻ đôi, nghe tim có bất thường...
Khi trẻ còn trong bụng mẹ có thể phát hiện bệnh qua siêu âm, nếu nghi ngờ sẽ tiến hành xét nghiệm nước ối. Người ta sẽ định lượng enzyme để phát hiện sự thiếu hụt enzyme 7-dehydrocholesterol reductase và xác định đột biến gen. Tuy nhiên những xét nghiệm này mới chỉ thực hiện được ở một vài phòng thí nghiệm ở các nước tiên tiến. Sau khi đứa trẻ ra đời nếu có nghi ngờ bệnh sẽ được xét nghiệm sinh hóa, định lượng lượng cholesterol máu (LDL cholesterol), sterol trong máu, phân tích đột biến gen, phân tích các men, xét nghiệm tế bào học... tất nhiên phải thực hiện các xét nghiệm chẩn đoán hình ảnh đối với các cơ quan bị dị tật.
Các biện pháp điều trị bệnh Hội chứng Smith Lemli Opitz
Không có điều trị đặc hiệu cho hội chứng và điều trị hội chứng Smith Lemli Opitz chủ yếu nhằm cải thiện triệu chứng và sửa chữa các khiếm khuyết phát sinh từ rối loạn này.
Bệnh nhân sẽ được đánh giá chặt chẽ bởi một nhóm các chuyên gia phối hợp hầu như tất cả các chuyên ngành. Bệnh nhân sẽ được theo dõi các khiếm khuyết ở tim, mắt, thần kinh, thần kinh cơ, tiêu hóa, tiết niệu sinh dục và lên một kế hoạch điều trị phù hợp.
Phẫu thuật có thể được chỉ định để sửa chữa sứt môi hoặc dính các ngón chân và các ngón tay. Một số dị tật tim cũng có thể được điều trị bằng phẫu thuật. Các bất thường về bộ phận sinh dục ở nam giới mắc hội chứng Smith-Lemli-Opitz cũng cần phải được phẫu thuật.
Trong một số trường hợp, bổ sung cholesterol có thể được bác sĩ khuyến khích để nâng cao tốc độ tăng trưởng cũng như tình trạng chung của bệnh nhân.
Ngoài ra còn có thể bổ sung thêm hormon bị thiếu, trợ thính, ăn chế độ ăn giàu cholesterol.
Đối với những trường hợp nhạy cảm với ánh nắng mặt trời, bệnh nhân cần sử dụng kem chống nắng, kính râm và quần áo phù hợp khi ra ngoài trời.
Do đây là 1 bệnh di truyền nên không có cách dự phòng hữu hiệu nào ngoại trừ việc làm tốt công tác sàng lọc trước sinh. Các cặp vợ chồng cũng cần tìm hiểu kỹ các thông tin về bệnh trước khi có ý định sinh con. Và cuối cùng là các cặp vợ chồng đã có 1 con bị hội chứng Smith Lemli Opitz thì không nên sinh tiếp nữa vì có 25% nguy cơ đứa con tiếp theo cũng sẽ mắc phải hội chứng này.